
导读
在原料药API的清洁验证中,对于残留物的接受标准的制定,行业里熟悉的是1/1000剂量或者是10ppm的标准,那么用PDE值(允许日暴露剂量)计算接受标准是必须的吗?
其实对于原料药的企业来说,问题很多时候不是不愿意用PDE值,而是PDE值的获取和计算是有相当大难度的。近期,一家欧洲的制药公司就对这个问题就提出困惑,因公司每一个药物/物质(substance)的PDE值需付费购买,而该公司就有超过200多个的药物/物质,整体来说是比较大的一笔费用。那么,该公司质量人员就提出,在API的行业中,清洁验证的限度是必须使用PDE值计算的吗?以及官方对这个问题的考虑和态度是如何的呢?
上一次主要分享欧洲方面的要求,本期补充中国和美国官方对API清洁验证中残留限度部分的要求。
01 中国篇
首先我们来看一下,中国GMP (2010版)附录《原料药》第24条,关于清洁验证的要求:
“应当根据溶解度、难以清洁的程度以及残留物的限度来选择清洁参照物,而残留物的限度则需根据活性、毒性和稳定性确定。”
“残留物的限度标准应当切实可行,并根据最有害的残留物来确定,可根据原料药的药理、 毒理或生理活性来确定,也可根据原料药生产中最有害的组分来确定。”
除此外,在中国GMP (2010版)附录《验证与确认》,第42条:
“活性物质残留限度标准应当基于毒理试验数据或毒理学文献资料的评估建立。”
另外,在2023年3月CFDI发布的《药品共线生产质量风险管理指南》中第3.1部分指出:
“…在评价 HBEL 时,每日允许暴露量(Permitted Daily Exposure, PDE)及每日可接受暴露量(Acceptable Daily Exposure, ADE)是被普遍接受的标准…”
备注:
具体计算公式见下图片:


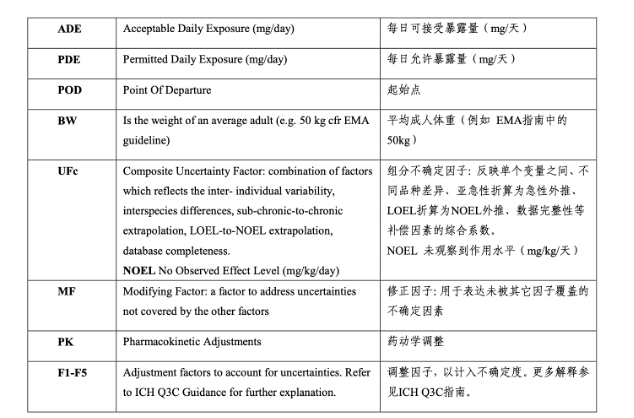
在4.2.3中原文指出:“相对于传统方法(如1/1000 最低日治疗剂量、10ppm 方法等)设定的限度来说,基于健康的暴露限度(HBEL)的可接受标准(如 PDE 值)在评估清洁残留数据时更具有科学性和优势…”
“…生产企业从传统的方法转为基于健康的暴露限度(HBEL)或其他毒理学数据评估时,当PDE值高于历史限值时,可选择继续采用历史限值…”,“…如果PDE方法计算出的限值低于历史残留限值,则应当使用基于PDE的新残留限度…”
在药品GMP指南(第2版,2023年发布),指出:
“对于具有活性的目标残留物,在可以获得足够数据时,应采用基于健康的暴露限计算MACO。或者也可采用10ppm或更低的固定值同步计算,然后取更低者。”
总结:实际上,对国内的相关GMP条款、GMP指南和指导原则进行收集整理后,会发现,对API工厂的清洁验证的残留限度的计算,PDE值也是作为推荐选项。但当不选择HBEL作为判断依据的时候,那么是建议工厂对选择的依据和标准做出分析和记录,说明背景原因。
02 美国篇
同样,我们先梳理的法规及指南分析美国官方的倾向。
In 21 CFR 211.67(a)
“Equipment and utensils shall be cleaned, maintained, and, as appropriate for the nature of the drug, sanitized and/or sterilized at appropriate intervals to prevent malfunctions or contamination that would alter the safety, identity, strength, quality, or purity of the drug product beyond the official or other established requirements.”
“设施和器具应进行清洁、维护,并根据药品的性质,在适当的时间间隔内进行消毒和/或灭菌,以防止发生故障或污染,从而改变药品的安全性、特性、效力、质量或纯度,使其超出官方或其他既定要求。”
In GUIDE TO INSPECTIONS VALIDATION OF CLEANING PROCESSES (1993), Chapter Ⅴ, link
“FDA does not intend to set acceptance specifications or methods for determining whether a cleaning process is validated..”
“FDA不计划制定验收标准或方法来确定清洁过程是否经过验证。”
“The firm's rationale for the residue limits established should be logical based on the manufacturer's knowledge of the materials involved and be practical, achievable, and verifiable.”
“公司制定残留限度的理由应该是合理的,是基于制造商对所涉及物料的知识并且是实际的,可实现的和可验证的。”
“Some limits that have been mentioned by industry representatives in the literature or in presentations include analytical detection levels such as 10 PPM, biological activity levels such as 1/1000 of the normal therapeutic dose, and organoleptic levels such as no visible residue.”
“业界代表在文献或报告中提到的一些限值包括分析检测水平(如10 PPM)、生物活性水平(如正常治疗剂量的1/1000)和感官水平(如无可见残留)。”
“Unlike finished pharmaceuticals where the chemical identity of residuals are known (i.e., from actives, inactives, detergents) bulk processes may have partial reactants and unwanted by-products which may never have been chemically identified. In establishing residual limits, it may not be adequate to focus only on the principal reactant since other chemical variations may be more difficult to remove.”
“与已知残留物化学特性的成品药物不同(即,从活性物、非活性物、洗涤剂),粗品工艺可能有没有经过化学鉴定的部分反应物和不需要的副产物。在确定残留限量时,只关注主要反应物可能是不够的,因为其他化学变化可能更难去除。”
总结:从上面可以看到,FDA对PDE或者说HBEL这个词没有在相关文件中进行提及,我们也收集分析了历年的警告性,也没有看到直接因未计算PDE相关而引发的问题。但FDA通常会对相关的问题,索引到SISQP(safety, identity, strength, quality, or purity of the drug product)原则性条款上。
备注:

