
400-166-2002
#技术转移 #注册申报 #药品上市 #化学药品 #治疗用生物制品
随着中国药品监管政策的不断开放与国际接轨,国家药品监督管理局(NMPA)出台了一系列政策,鼓励境外已上市药品通过技术转移在境内生产,以提高药品的可及性和满足公众的用药需求。政策旨在通过简化药品注册申报流程,降低境外药品境内生产的技术壁垒,同时确保药品质量和疗效与原产国保持一致。政策涵盖了GMP要求、药品注册审评审批制度的改革,以及对境外药品生产企业的监管合作等多个方面。
接下来,我们一起来看看相关政策和对比已上市境外生产转移至境内生产的化学药品和治疗用生物制品征求意见稿和正式稿的变化。
01 相关法规
2021年1月12日,NMPA发布了《药品上市后变更管理办法(试行)》,旨在进一步规范药品上市后变更,加强药品监管部门药品注册和生产监督管理工作的衔接。
其中第十条规定:“已在境内上市的境外生产药品转移至境内生产的,应当由境内申请人按照药品上市注册申请的要求和程序提出申请,相关药学、非临床研究和临床研究资料(适用时)可提交境外生产药品的原注册申报资料,符合要求的可申请成为参比制剂。具体申报资料要求由药审中心另行制定。”
附件3政策解读中第八条提到,境外生产药品(原进口药品)通过药品生产技术转让为境内生产的,境内持有人可在2年的过渡期内继续按518号文的要求办理。
2023年03月23日CDE发布了《已在境内上市的境外生产药品转移至境内生产药品上市注册申请申报资料要求(化学药品)(征求意见稿)》及相应的政策解读。
2023年12月22日CDE发布了《已上市境外生产药品转移至境内生产的药品上市注册申请申报资料要求(治疗用生物制品)(征求意见稿)》。
2024年05月09日CDE发布了《已上市境外生产药品转移至境内生产的药品上市注册申请申报资料要求(化学药品)》通告。2024年第22号
2024年06月14日CDE发布了《已上市境外生产药品转移至境内生产的药品上市注册申请申报资料要求(治疗用生物制品)》的通告(2024年第30号)
02 政策变化
小编将征求意见稿和正式稿进行对比,一起看看两者的政策变化吧。
①《已在境内上市的境外生产药品转移至境内生产药品上市注册申请申报资料要求(化学药品)》
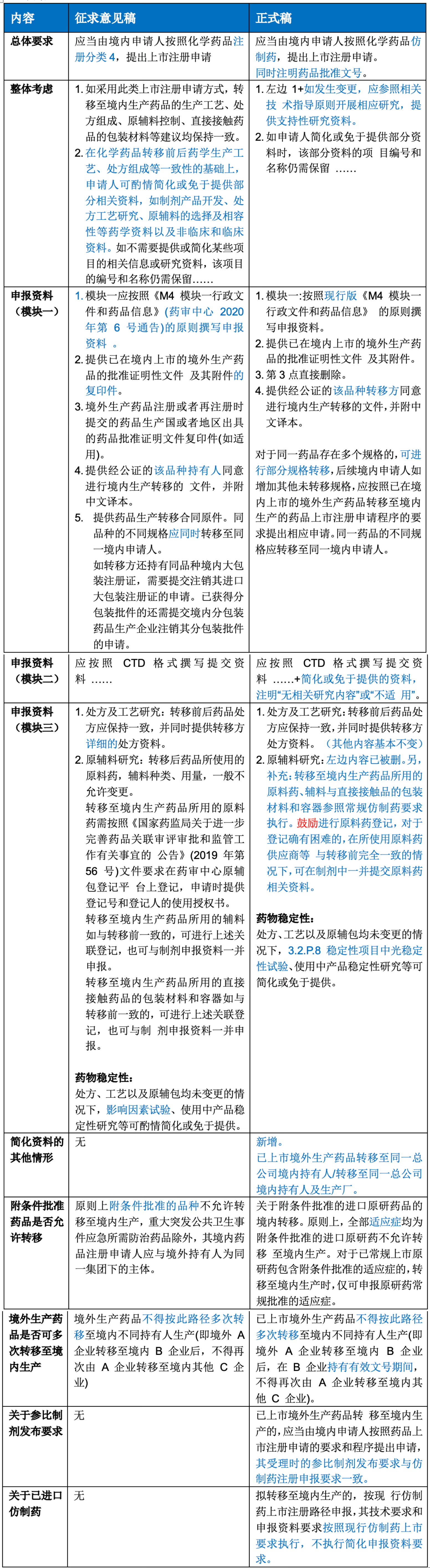
②《已在境内上市的境外生产药品转移至境内生产药品上市注册申请申报资料要求(治疗用生物制品)》

以上是本期分享的已上市境外生产移至境内生产的化学药品和治疗用生物制品关于上市注册申报资料的政策变化总结。
P.S. 本文章为gempex德恩咨询原创。如需转载,请注明来源于gempex德恩咨询。
欢迎垂询
服务热线:400-166-2002
邮箱:info-cn@gempex.com

