
400-166-2002
AI,GMP合规,GMP认证,GMP咨询,制药合规,制药行业,药品监管,质量管理,德恩咨询,gempex
一台AI驱动的智能灯检机正在药厂全速运转,每秒检测数百支安瓿瓶。但在不远处的会议室里,企业的质量负责人正面临监管人员的关键提问:“如何证明它今天的判断标准,与你们当初验证放行时完全一致?”
随着欧盟GMP附录22(人工智能)征求意见稿的发布,AI在制药领域的合规监管已从前瞻探讨进入实质性的框架构建阶段。对于行业而言,准确理解监管逻辑、建立清晰的AI认知并掌握关键控制点,已成为一项紧迫任务。
全球主要监管区域对AI在制药领域的应用已形成明确导向。一方面,各国通过战略规划积极引导技术应用(如中国《医药工业数智化转型实施方案》、美国相关行政命令);另一方面,具体的监管要求正迅速落地。其中,2025年7月欧盟发布的GMP附录22(人工智能)草案具有里程碑意义,它首次在GMP层面系统性地提出了对AI/ML的监管考量。
这意味着,AI在制药行业的应用将面临 “双重合规”要求:
企业需在两者交汇处建立起有效的管理体系。
有效的合规始于准确的技术理解。根据ISPE指南及欧盟附录草案,基于风险的分类管理是核心原则。
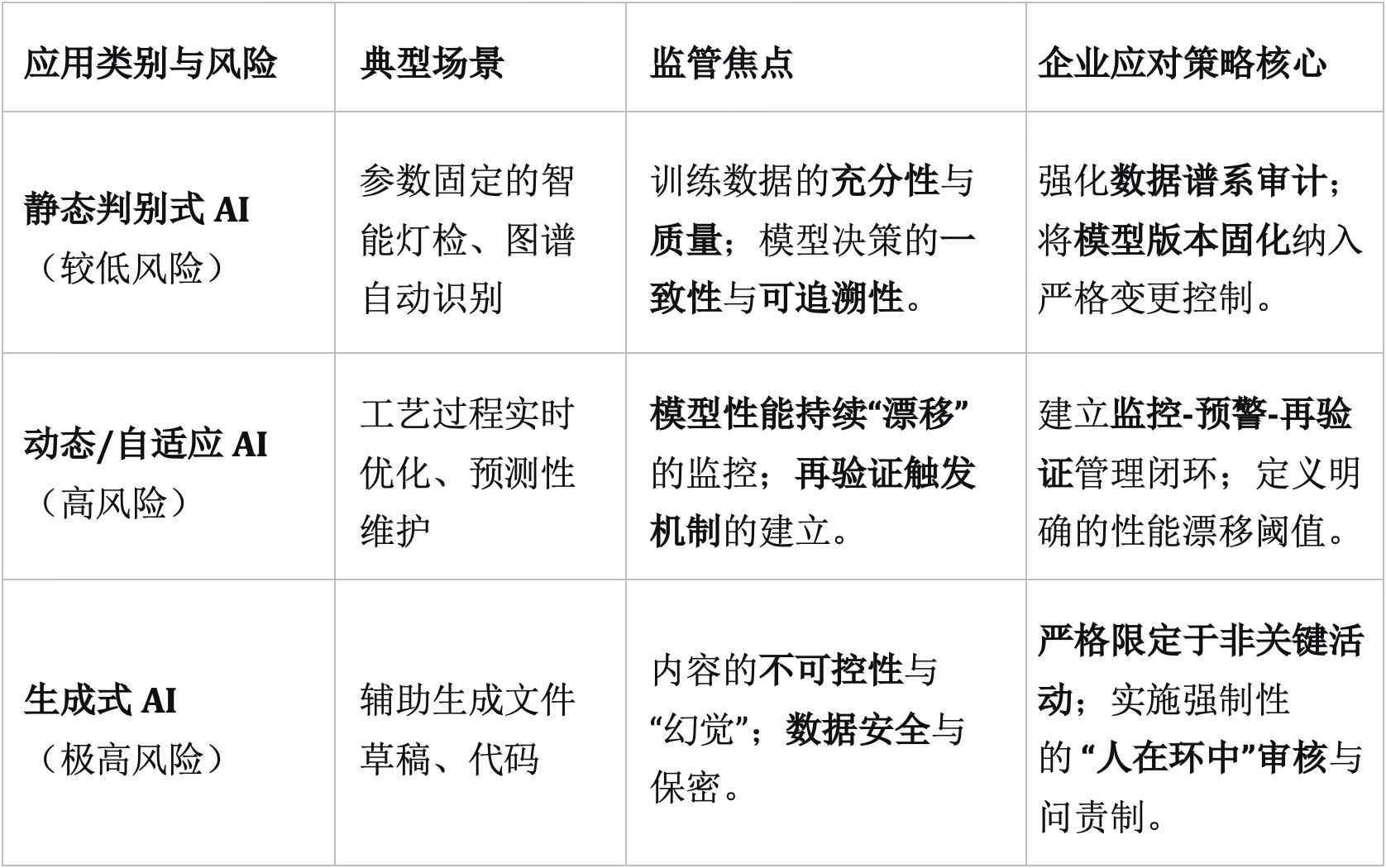
判别式AI:如同一个“专业鉴别师”,学习输入与输出的映射关系,用于分类或预测。例如分析医疗影像、检测产品外观缺陷。其特点是在限定数据范围内工作,不产生新数据,硬件需求相对较低,风险相对可控。
生成式AI:像一个“内容创造者”,学习数据分布并生成全新内容,例如撰写文本,生成代码或设计分子结构。它创造力强,但输出的不可预知性也带来了高风险。因此,当前监管态度明确:生成式AI目前仅能用于非关键的GMP活动,且必须采用“人机回环”模式,由经培训有资质的人员对最终输出负责。
静态训练模型:采用固定数据集进行一次性的训练,模型部署后参数不变,如传统的智能灯检机。其优势在于状态固化、易于验证,但无法适应新变化。
动态训练模型:持续加入新数据进行学习与优化。这虽能提升模型在变化环境中的表现,但也引入了 “模型漂移”(即性能随时间或数据变化而衰减)这一核心风险,使持续监控和再验证成为必须。全球生物制药行业合作组织BioPhorum在2025年7月发布的指南中,特别强调了应对数据漂移和模型可解释性的挑战。
将AI视为一个有生命的实体进行管理至关重要。其生命周期独立于其所嵌入的计算机化系统,涵盖从业务需求定义、数据收集处理、模型训练验证,到部署放行、运行监控、变更迭代直至退役的全过程。全球行业指南已开始将这一AI生命周期与经典的验证V模型进行对标,旨在为这一新事物找到与既有质量体系衔接的路径。
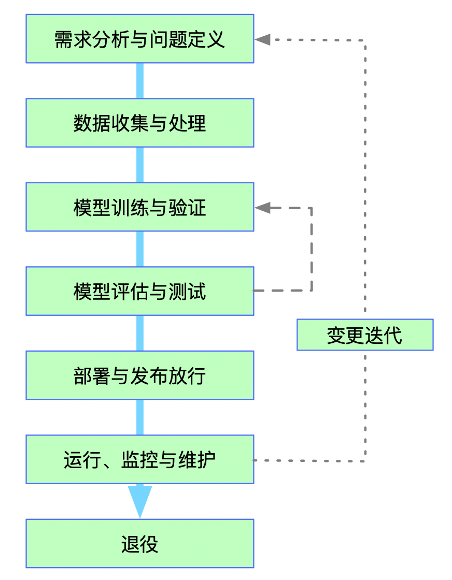
其中,数据管理是合规的重中之重。在AI项目中,数据不仅是记录,更是模型的“教材”和“燃料”。因此,数据完整性(ALCOA+原则)的监管范围发生了关键性延伸:
欧盟GMP附录22草案为不同类别的AI指明了差异化的控制要求,这为企业制定策略提供了直接依据:
一个务实的建议是:企业若使用生成式AI辅助生成文件(如SOP草案、验证报告),应明确界定其仅为提高效率的“工具”,所有输出必须由责任人员完成内容的实质性审核、批准与承担责任,而非直接定义AI参与了“非关键GMP活动”,以避免复杂的合规性争论。
面对清晰的监管信号,企业的行动路径应系统而务实:
第一步:全面盘点和风险分类
识别所有已用及计划引入的AI工具,依据其功能(判别/生成)与模式(静态/动态)进行风险分级。
第二步:开展差距分析与策略制定
以欧盟附录22、FDA指南等为基准,评估现有实践差距,并针对高中低不同风险制定差异化的验证与监控策略。
第三步:升级质量体系与实施管控
将AI治理要求嵌入现有体系。例如,在供应商审计中增加对AI开发生命周期的审查;在用户需求中明确要求可解释性;在变更控制程序中新增“模型迭代”类别。
相关专家介绍
如果您想了解更多GMP解决方案,可以访问 www.gempexchina.com/gmp-knowledge 获取。在那里,您将更深入了解gempex德恩咨询的GMP服务。
如果您有具体的合规难题,请通过以下方式联系我们:
热线:400 166 2002
gempex德恩咨询深耕GMP合规领域23年,致力于为全球的生命科学企业提供合规、高效及可执行的GMP解决方案,帮助制药、生物技术、原料药、化学、医疗器械、原辅包和化妆品等各方达到GMP标准,减少合规及药品安全风险。
目前,我们拥有60多位经验丰富的GMP专家,全球累计执行项目超过5000个,累计为1000多个客户提供专业服务,业务遍布20多个国家,并与众多知名药企建立了长期的合作关系。

