
# 新建工厂 #GMP指导
新建厂房雷区
回想以上 “新建厂房雷区” 是不是心有余悸?
今天,小编整理了8条关于新建工厂的常见问答知识,希望对小伙伴们在新建工厂需求方面有所帮助!
Q: 我司准备新建一个制药工厂,目标是中美双报。请问GMP的相关工作在什么时间介入是及时且有效的,以便不会影响后续市场?
A: 德小恩的一句话建议当然是越早越好!如需要第三方协助尽量在厂房设计阶段的前期就确认好合作服务商。
我们都知道GMP的核心就是防止药品生产中的混批、混杂、污染和交叉污染。因此,在厂房设计方面需要综合考量很多因素,包含从厂址选择和厂区总体布局,以及生产区、包装区、储存区、质量控制区、辅助区、人流和物流等区域,通过质量法规风险和技术风险的评估,设计和实施GMP相关规范条款[1-2]。
举例美国FDA在厂房设计和建造以及无菌厂房设计安排方面,就有规定:
1. 美国FDA CFR 211 Subpart C-Buildings and Facilities 中Sec.211.42 design and construction features写明[3]:
任何用于药品生产、加工、包装或贮存的厂房或建筑物,大小适宜,结构与位置使其易于清洁、保养且适合操作。
厂房或建筑物有足够空间来有条理地安装设备和放置材料,避免不同类的成份、药品容器、密封件、标签、中间体或药品等相互混放,防止污染。通过厂房的上述物料其流向在设计时要防止污染。
2. 美国FDA Sterile Drug Products Produced by Aseptic Processing IV. BUILDINGS AND FACILITIES中E. Design部分中对于无菌厂房的设计安排。摘取几点内容:设计无菌操作过程是为了减少无菌药品在生产操作过程中暴露受污染的危险。减少无菌成分的暴露时间,提供最大可能的环境控制,优化生产流程,设计设备以防止低质量的空气进入100级(ISO 5)的洁净区,这些过程对获得高无菌保证非常关键[4]。另外,应当优化人员和物料流程以减少不必要的活动,防止增加对暴露产品,容器或周围环境的潜在污染的可能。同时,也要求进入传统洁净室或隔离箱等关键区域的次数要减少[4]。
而中国、欧盟和WHO的GMP也对于厂房设计也做了具体要求,具体可参考中国2010版GMP第四章-厂房与设施内容以及厂房设备GMP实施指南-厂房设计章节[1-2];欧盟可参考欧盟GMP第三章-厂房与设备内容[5];WHO GMP 12章厂房设施(Premises)也对厂房设计原则、一般性法规规、辅助区域、仓储区、称量区、生产区、质量控制区做了规定[6]。具体请查询文章内容!
A:设计审核要点是针对产品特点、厂区位置、生产区布局、公用系统、仓储区、质量控制区及其他辅助功能区等规划进行审核。如何将产品和法规的要求转化到工厂设计中,需要通过结合产品的性质、工艺路线、关键控制要求、生产方式、产能规划、法规要求等多方面的信息,系统的进行挑战和讨论,全面植入到工厂的设计中。
设计审核服务限于GMP符合性的审核,以及重点在GMP相关的区域和系统,非GMP相关的区域和系统只有在可能对GMP区域造成影响时,增加相关的影响性审核,例如,评估人流时,办公室位置的影响。
在设计审核过程中,德恩的相关审核流程如下(但不局限于):
收集文件及项目信息
首次审核关键文件
撰写报告
客户和专家答疑
图纸更新后的二次复核
如有,进一步的更新版图纸审核及讨论
图纸确认后的最终报告
其中,项目基本信息用于理解项目,包含:项目介绍、计划市场、产品信息、工艺流程,工艺描述,物料平衡等。
审核的关键文件如下(包含但不局限于):
基本图纸审核:工厂布局、楼层布局、设备布局图、洁净区域划分图、工艺路线和人流物流图、储存区、压差分布图、质量控制区等;
暖通空调系统:空调P&ID图、送风平面图、排风及回风平面布局图等;
管道系统:公用系统的分配系统P&ID图纸,工艺排水及地漏等;
......
Q:新建工厂过程中如何进行施工指导及技术转移指导?
A:施工阶段设计的设施设备如生产设备、空调系统、纯化水及注射用水、特殊气体及压缩空气等的安装和调试过程的良好GEP管理可有效促进验证确认的实施。我们建议确定设备及设施的验证策略,并根据施工进度建立涵盖GMP活动的全面计划,对施工过程进行规划。针对在施工过程中的GMP关键控制点,技术专家可以现场进行审核和指导。
对于需要进行技术转移的,应提早制定转移策略,并收集整理及分析:
物料与产品质量标准
物料产品检验方法
物料、产品分析方法及验证包
对匹配进行检查,转移风险进行评估
对工艺转移资料包符合性及完整性审核
生产及工艺转移过程的合规性等
Q: 文件体系几百个,人员少且紧张,如何快速有效的对文件体系进行搭建?
A: “一把抓,无头绪,人员未全部到位,我快要凌乱了…”。为了规避此种情况的发生,小编准备了2张关于“如何建立一个制药设施-GEP&GMP”基本流程图以及文件整合过程中的优先顺序图及工作整合图,希望对您的文件准备工作有所帮助!
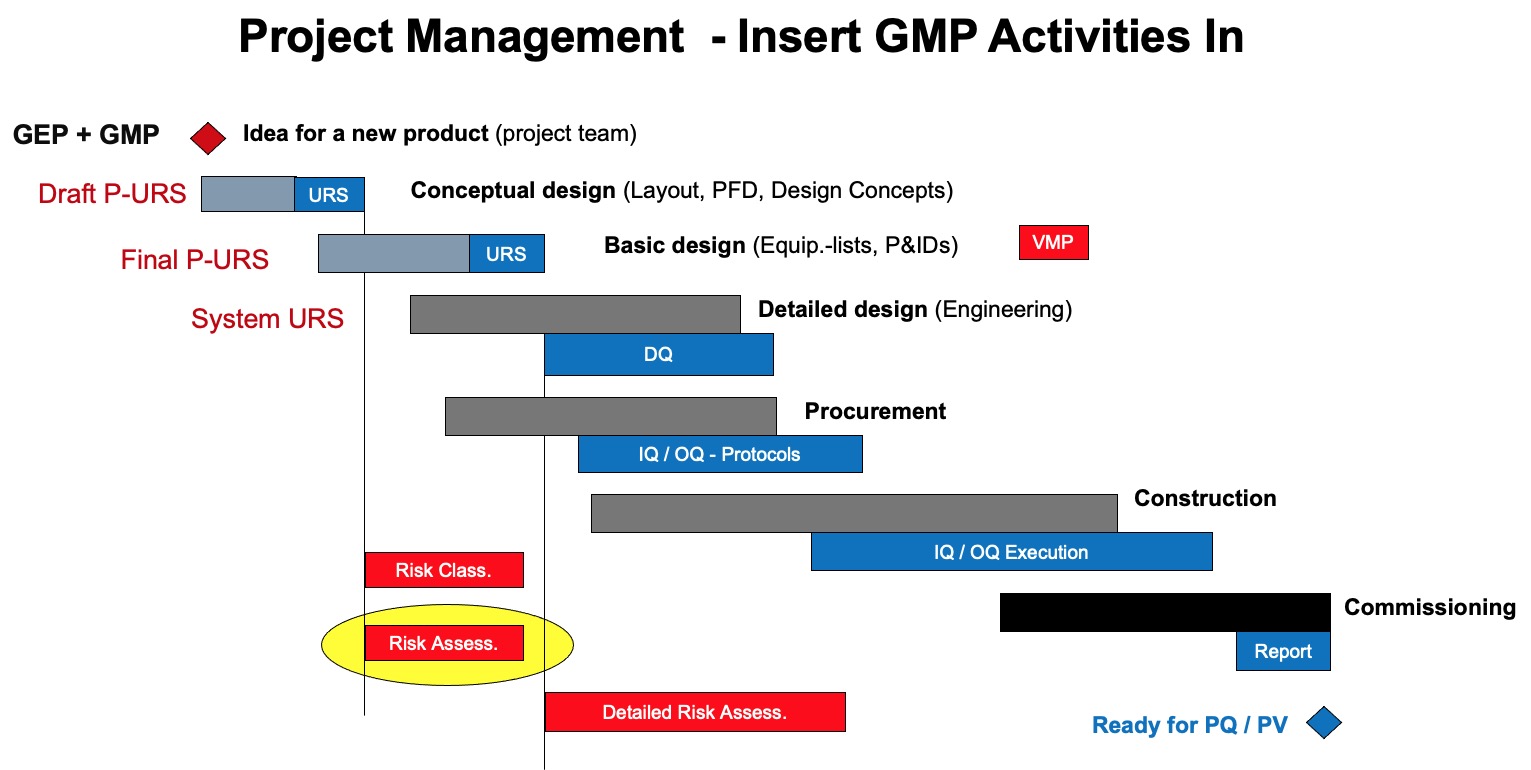
图1:新建药厂基本流程图
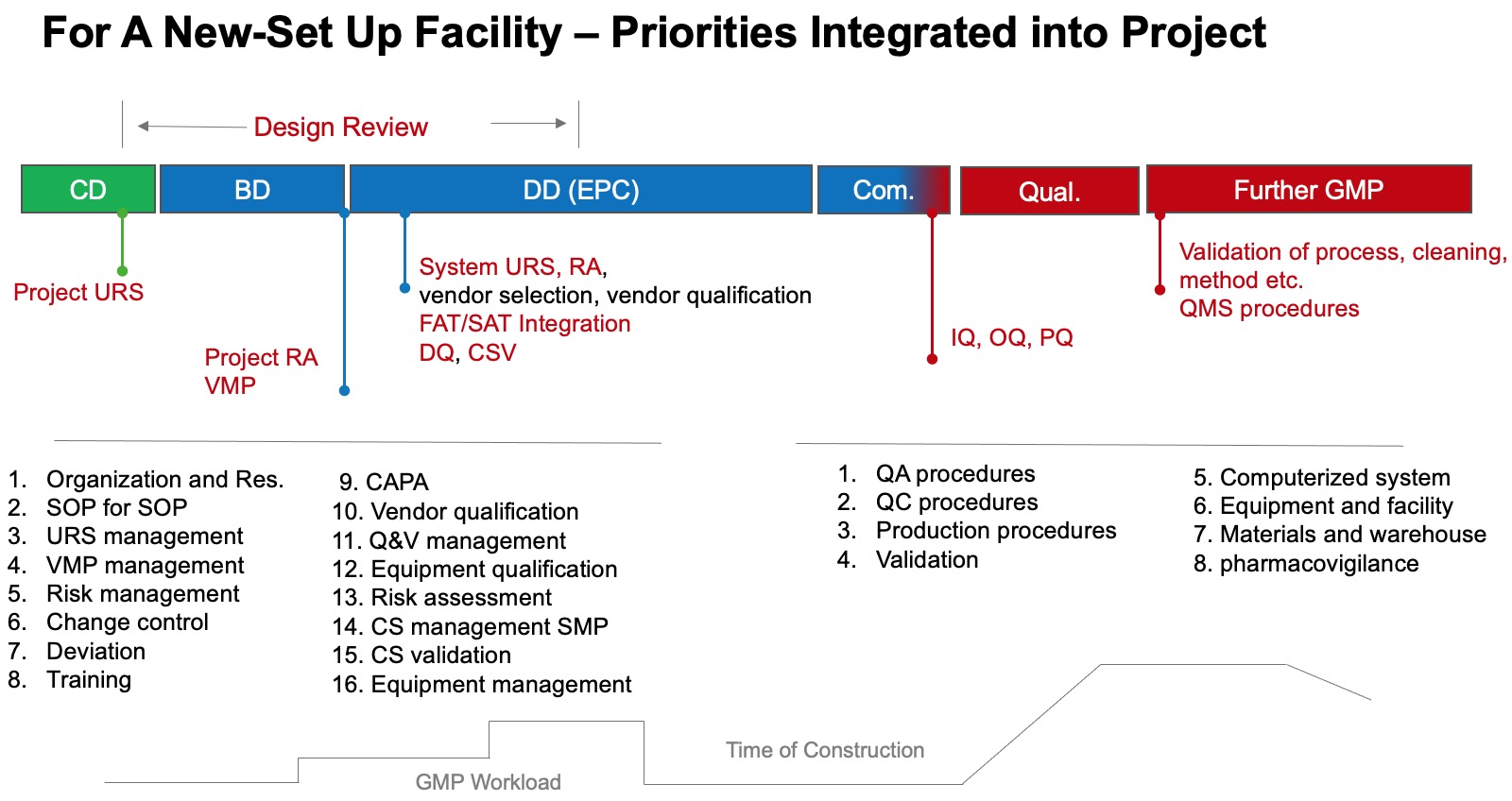
图2:新建工厂-文件整合优先顺序
这里,德小恩建议您参考上述流程图,将文件划分不同的优先等级,一般划分为若干个阶段来完成。
第一阶段优先建立(较紧急)如:组织架构、文件管理、验证确认管理等;
第二阶段根据项目进度安排次要紧急的文件,以此类推。
Q: 智能化是我司的特色之一,如何做好计算机化系统合规性以彰显我司特色?
A:整个完整的计算机化系统验证过程,是遵循系统整个生命周期全过程的,涵盖从概念、项目、运行到退役。德恩计算机化系统合规性包含:系统体系搭建、系统验证、软件验证、基础设施平台确认。详细可以参考以往德恩微信公众号发布有关计算机化系统文章。
Q: 如何进行设备设施以及工艺/清洁确认验证?
A:一个高效的验证确认理念,能够帮助企业在符合性的基础上节省更多的时间和资源,确保项目的有序推进。
1. 设施设施
首先需要根据目标市场的法规要求对设施设备实施验证确认以确保满足工艺要求。设备确认验证的主要内容及工作步骤:
a. 依据设备清单,进行分析哪些是需要进行验证的设备清单目录,哪些是不需要验证确认的设备(需要校验的,请送有资质的校验公司实施)。
b. 对需要验证确认的设备进行分类。如标准设备(如安全柜、冰箱、发酵罐等),采用简化的验证确认策略(如URS及风险评估合并,DQ/IQ/OQ/PQ(如有)合并编写;若为定制化或复制的设备(如空调系统、洁净室、纯化水系统、注射用水系统等)将采用相应的验证确认策略,如独立的URS和风险评估等。
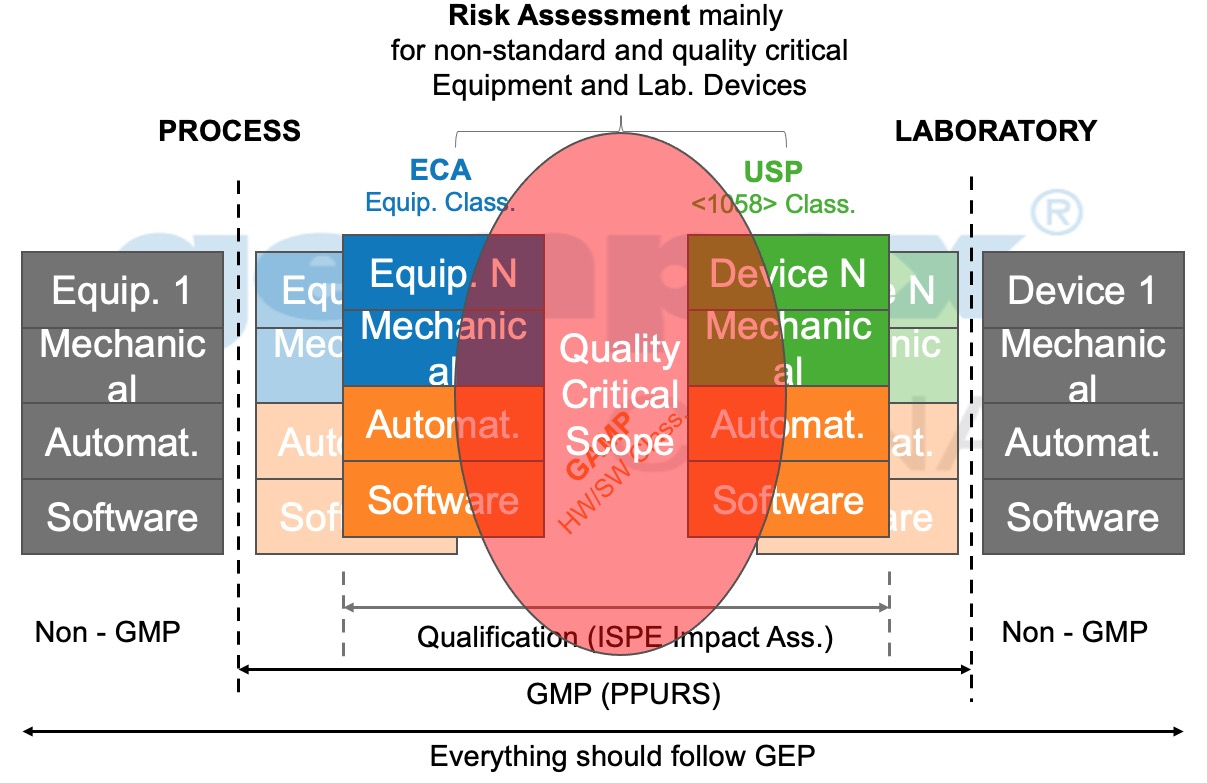
图3:风险评估
2. 工艺验证
GMP对工艺验证的要求贯穿于工艺的整个生命周期。对于药品包含新公司的初次验证、工艺变更后的验证、现场转移及持续的工艺验证。工艺验证需要证明是否所有的质量属性和工艺参数(被认为可以确保验证状态和产品质量的重要项目)能够与工艺持续相符。明确关键工艺参数、关键质量属性和相关的可接受标准[7]。特别应注意新的验证理念的图示:
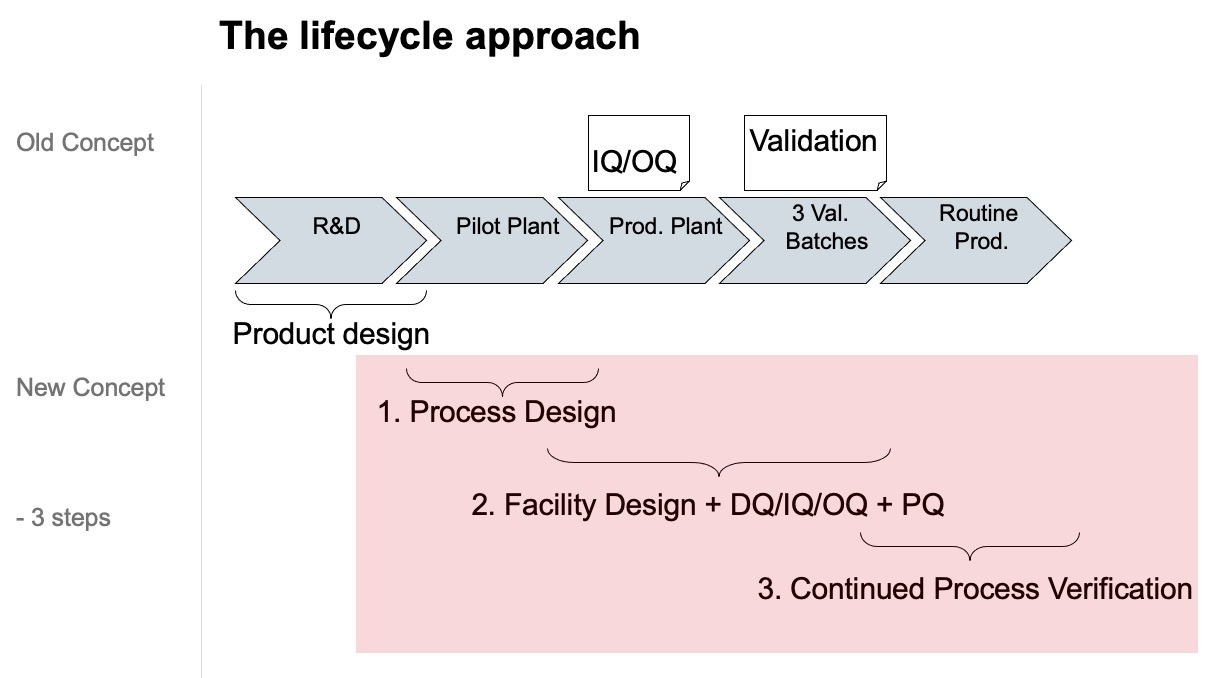
图4: 新的验证理念
3. 清洁验证
首先,清洁验证具体法规要求可参考中国GMP附录-确认与验证[8]以及欧盟GMP《确认与验证》附录内容[7]。
其次,清洁验证评估是清洁验证中较难的一个环节,只要完成清洁评估报告,清洁验证方案的编写就显得比较容易。为了清洁验证评估的编写更清晰和更有逻辑,德恩概括了清洁验证评估报告内容如下图:

图5: 清洁验证评估报告内容
Q: 整个过程,都需要哪些人员或者部门参与?
A:德恩建议配备关键对接人,同时至少需要包括工程、质量及验证人员参与。其余的工作可交付德恩来协助支持。
Q: 德恩作为专业的GMP咨询服务商,在新建工厂项目上,可以提供哪些服务以及成功案例?
A:德恩可以提供新建工厂的全生命周期GMP咨询和执行服务,根据项目的推进可分为项目前期、中期和后期,具体包含:
前期阶段:GMP项目管理、项目URS编写及风险评估、设计审核、VMP编写等
中期阶段:各系统的URS、风险评估、FAT/SAT的整合、供应商评估和筛选、设备及公用系统的确认、计算机化系统验证、质量体系等工作的审核、编写、执行或协助
后期阶段:验证类工作如工艺验证、清洁验证、分析方法验证等方案编写或审核,生产许可证事项申报支持、质量体系运行服务如培训、内审、批记录审核、体系执行工作、官方检查陪同等。
表一:德恩战略长期合作成功案例(选取部分)

以上是本次分享的8个新建工厂常见问答知识。最后希望大家都能顺利圆满的完成一个新建工厂的各项工作,成功通过检查产品上市,但是要记住哟:
Key Concept for Pharmaceutical Project
Pharmaceutical projects are complex due to the numerous regulatoryand technical requirements and the many disciplines involved
From many past cases you know, to keep timelines and budgets expects a detailed planning and an early start of all relevant GMP activities
参考资料:
[1] 中国药品生产管理规范(2010年修订)及相关附录.
[2] 中国2010版厂房设备GMP实施指南.
[3] US Food and Drugs Chapter I-Food Drug Administration Department of Health and Human Services Subchapter C-Drugs: General Part 211 Current Good Manufacturing Practice for Finished Pharmaceuticals.
[4] Guidance for Industry Sterile Drug Products Produced by Aseptic Processing IV. BUILDINGS AND FACILITIES. FDA. September 2004. Pharmaceutical CGMPs.
[5] EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use Part1 Chapter3: Premises and Equipment.
[6] WHO Good Manufacturing Practice for Medicinal Products:main principles Annex2.
[7] EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use Annex 15: Qualification and Validation.
[8] 《药品生产质量管理规范(2010年修订)计算机化系统和确认与验证附录公告(2015年第54号).国家药品监督管理局.2015.5.26.
[9] Good Manufacturing Practice-Medicinal Products for Human and Veterinary Use: Annex 11: Computerised Systems. The Rules Governing Medicinal Products in the European Volume 4.

