
400-166-2002
GMP法规,GMP合规,GMP认证,GMP咨询,制药合规,制药行业,药品监管,质量管理,德恩咨询,gempex,FDA483,FDA,EIR,warning letter,untitled letter
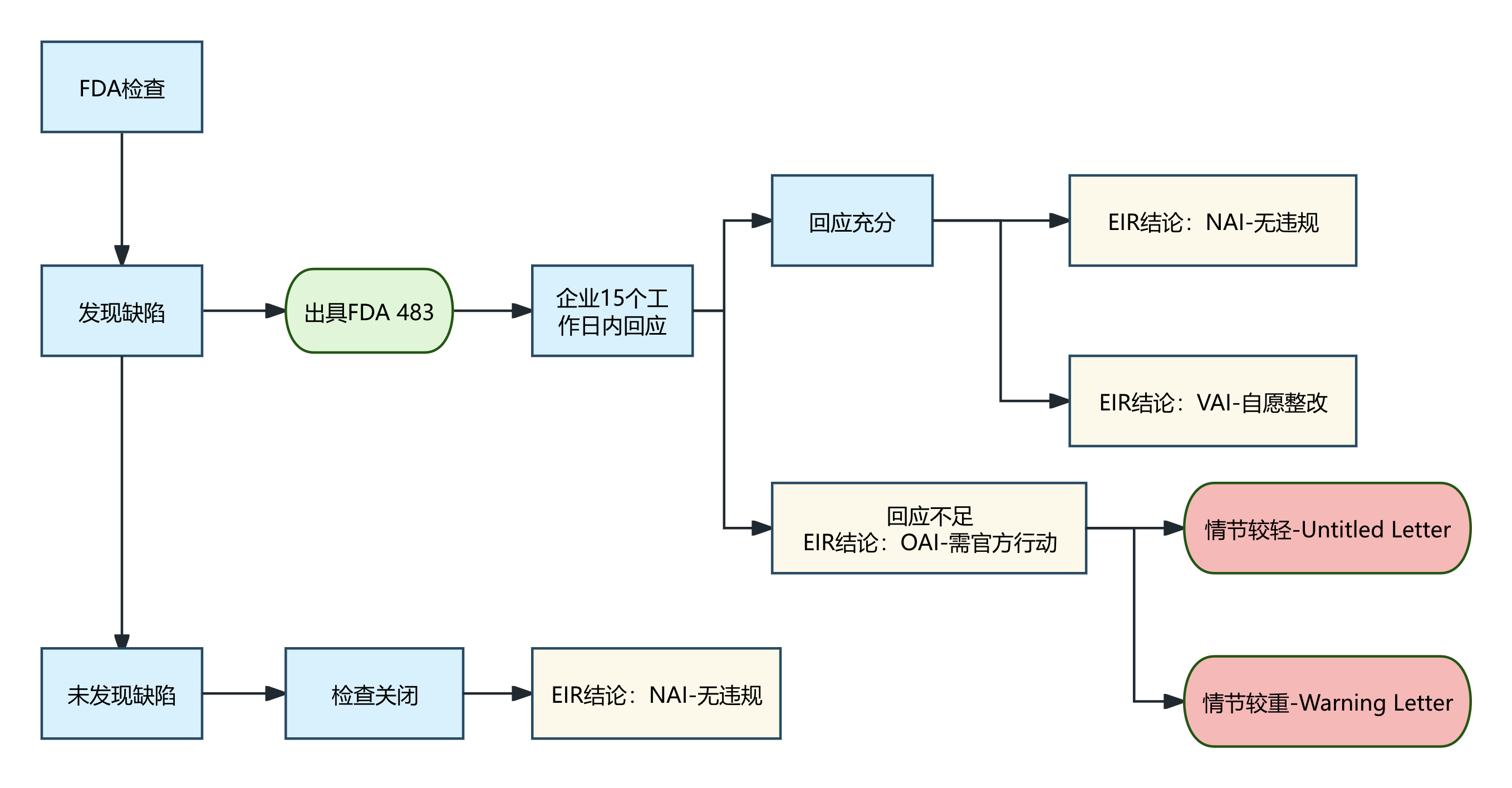
在FDA的监管体系中,有四份文件构成了从问题发现到检查终结的完整路径。
它们是哪四份文件?
1、FDA Form 483(现场检查缺陷表):检查结束后,检查官当场出具的观察项清单,企业需在15个工作日内作正式回应。
2、EIR(机构检查报告):检查员在现场检查结束后30个工作日完成并提交EIR初稿给地区办公室甚至中心(如CDER或CBER)审核,审核通过后,发给企业盖章,代表此次检查的最终监管结论。
a)包含了检查细节、调查官对483观察项的结论,以及FDA总部的最终结论:NAI-无违规、VAI-自愿整改、OAI-官方行动。
b)任何FDA现场检查完成后都会生成此报告。
3、Untitled Letter(无标题信):一种较轻的官方警告。针对的违规行为不涉及《联邦食品、药品和化妆品法案》的核心原则,但需要纠正。
4、Warning Letter(警告信):FDA针对严重或系统性违规发出的最高级别执法警告。
它们核心区别是什么?
|
文件 |
法律性质 |
是否主动公开 |
核心目的 |
|
FDA Form483 |
非正式观察记录 |
不主动公开 |
启动整改流程 |
|
EIR |
内部行政结论 |
不主动公开 |
给出检查结论 |
|
Untitled Letter |
正式监管告知 |
FDA官网可能公开 |
要求纠正违规 |
|
Warning Letter |
正式执法警告 |
FDA官网公开 |
警告法律后果 |
如何应对这些监管文件?
总结
EIR是整个监管流程的枢纽。企业在483阶段的回应质量,直接决定了EIR的结论,进而影响是否会收到正式信函。最经济的策略是:在483阶段投入资源做好根本原因分析和彻底整改,争取良好的EIR结论,从而避免进入更严肃的正式监管程序。
应对FDA检查是一项系统工程,从483的即时响应、CAPA计划的制定,到应对正式信函的策略沟通,都需要深厚的法规理解和实战经验。
gempex德恩咨询作为源自德国的GMP咨询与执行机构,拥有超过20年的行业经验,在FDA合规与审计应对领域拥有丰富经验,我们的专家团队可为您提供:
如果您的企业在应对FDA合规挑战或现场迎检方面需要支持,欢迎联系我们。
如果您想了解更多GMP解决方案,可以访问www.gempexchina.com/gmp-knowledge获取。在那里,您将更深入了解gempex德恩咨询的GMP服务。
如果您有具体的合规难题,请通过以下方式联系我们。
热线:400 166 2002
邮箱:info-cn@gempex.com
关于gempex德恩咨询
gempex德恩咨询深耕GMP合规领域24年,致力于为全球的生命科学企业提供合规、高效及可执行的GMP解决方案,帮助制药、生物技术、原辅包、医疗器械等各方达到NMPA、EU、FDA、PIC/S等GMP标准,减少合规及药品安全风险。
目前,我们拥有60多位经验丰富的GMP专家,全球累计执行项目超过5000个,累计为1000多个客户提供专业服务,业务遍布20多个国家,并与众多知名药企建立了长期的合作关系。

