
GMP法规,GMP合规,GMP认证,GMP咨询,制药合规,制药行业,药品监管,质量管理,德恩咨询,gempex,清洁验证
【引语】
“目视检查能否作为清洁验证的唯一可接受标准?”——这个话题不仅在行业里热议不断,在我们团队内部,两位技术专家也曾为此展开一场激烈的“神仙打架”。一方坚守法规红线,另一方则深挖科学内涵。
本文将这场精彩的内部讨论整理成文,带你深入剖析EMA、FDA、NMPA等监管机构的指南深意,并介绍ASTM标准提供的系统评估工具。无论你是急性子想直接看结论,还是喜欢跟随专家的思路一探究竟,这篇文章都能给你带来启发。
【太长不看版 | 核心观点速览】
1.原则上不行: 根据NMPA、EMA等主流法规,目视检查通常不能作为清洁验证的唯一标准。
2.理论上有例外: 法规留了口子(如专用设备、低风险场景),但条件极其严苛。
3.落地有工具: 想用这个“例外”,必须通过科学评估。比如ASTM E3106-22标准,它要求综合评估清洁工艺的重现性、残留物的毒性、目测方法的检出限 三个维度。
4.检出限需验证: 目测不是看一眼那么简单,其方法本身需要验证(参考ASTM E3263-22),且其检出限应低于基于健康暴露限度(HBEL)计算出的残留限度。
5.最终建议: 对多数企业而言,直接取样检测可能更稳妥。但对于产品复杂、资源有限的原料药(API)企业,这套评估工具能帮助识别真正的高风险点,实现资源优化配置。
【完整讨论】
今天我们来聊一个在制药行业里“老生常谈”却又“屡吵不爽”的话题:清洁验证中,能不能只用“目视检查”作为唯一放行标准?
A:都2026年了还在讨论这个?这个话题还用聊吗?目视检查肯定不能作为唯一放行标准,NMPA《清洁验证技术指南》法规明确“通常不能作为单一可接受标准使用”。
B: 法规是“通常不能”,并非“完全不能”,它并没有一刀切地完全禁止。像EMA附录15的正式稿用的是“generally not accepted”,也是没有一刀切。“generally”这个词不是随便写的,它是当年行业与监管机构博弈的结果,为一些特定情况留下了豁免的空间。以下截图是EMA附录15征求意见稿关于目视检查的描述,可见在征求意见稿中是没有“generally”一词的。

从其他国家的法规指南看,也有保留豁免空间。
这说明监管机构认可在特定场景下的简化处理。
A:就算法理上留个豁免,但法规和监管本身都没有给出“只用目视检查作为唯一放行标准”这种例外的具体说法。
B:关于例外,如果只考虑化学残留的情况下,你觉得哪些场景可能只靠目视就够了?
A:我想到API的前端工序,法规本身就有弹性空间;或者制剂里那些只接触一种低风险辅料的简单设备,例如一把专用的称量勺,它又好洗又好查,风险可控。
B:说到点子上了!这些例子的共性就是风险双低:残留物的毒性低,设备清洁检查难度低。这正好呼应了EMA在2018年关于HBEL指南的问答文件,在问题7和问题8(以下简称Q7和Q8)的回答中给出的评估目视检查的几个维度。
关于HBEL指南的问答文件:即EMA《交叉污染和共用设施清洁限度指南问答》。
HBEL:Health-Based Exposure Limit 基于健康的暴露限度,用于衡量API不会对健康造成危害的最大安全剂量,它是药企进行共线生产(多款药在同一车间生产)和清洁验证的“金标准”。
A:但Q7所提到的目测是在完成清洁验证后的日常目测,不是清洁验证中的目测,这在今天讨论的主题中不具备直接参考意义吧?
B:确实,Q7的前提是已完成清洁验证。但它们逻辑是相通的,监管机构在这里给出了评估的方向:当你想用目测证明清洁度时,必须考虑:
1. 清洁工艺的重现性;
2. 残留物的危害性;
3. 目测方法的检出限是否能满足通过HBEL算出来的限度。

A:有了方向,但具体如何评估呢?这个问答并没有给出详细的评估工具。没有工具,意味着企业就算考虑应用目视检查,也会感到无从下手,难以操作。
B:你点出了痛点,针对这一“工具空白”的难题,美国材料与试验学会(以下简称ASTM) 提供了一份标准文件——E3106-22:《基于科学和风险的清洁工艺开发和验证标准指南》。
这份标准文件用于指导制药和医疗器械企业如何科学地清洗生产设备,它吸收了行业内组织机构做的研究成果,形成了一套系统的清洁验证风险评估工具,其中包括目视检查适用性的评估。
因此,企业可以参考这份标准,通过评估工具,实现从“方向”到“方案”的落地。
简单来说,这个评估工具用两个维度进行评估:
1.Cpu分数。这个Cpu不是我们熟知的电脑CPU,而是代表基于上限计算的过程能力指数(该指数为工艺验证和产品质量回顾领域常用,这里将其引入到了清洁验证领域)。
基于以往的残留检测数据,通过计算Cpu来评价清洁工艺本身的重现性或者能力。Cpu分数越低,代表清洁工艺能力越好。
2.毒性分数。即残留物本身的毒理学数据。国内比较贴近的概念是OEB。毒性分数越小,代表残留物毒性越低。
OEB:职业接触等级或职业暴露等级。如果把化学品或药物比作“危险分子”,OEB 就是给这些分子贴上的“危险程度标签”。当某种物质缺乏具体的法定安全浓度标准(OEL)时,OEB 就被用来快速评估其危害等级。
当Cpu分数和毒性分数都较低时,ASTM E3106-22建议可以考虑在新产品对应设备/工序的清洁验证中仅使用目测。
A:但这个的前提是,要有数据计算Cpu。如果是新工厂新设备,就没有任何历史数据来计算Cpu。在这种情况下,不同产品切换之间的清洁方法,还能在清洁验证中使用目测作为唯一标准吗?
B:在没有任何数据作支持下,对于不同产品切换之间的首次清洁验证就仅用目测,当然是不可以的。据我所知,2025年2月份FDA就给没有提供清洁验证检测数据的企业开过警告信。
A:我发现这个标准并没有回答我们之前讨论的Q7的第三个评估方向——目测方法的检出限是否能满足通过HBEL算出来的限度(即目测方法的检出限应该如何确认)。
B:E3106-22标准本身更多还是聚焦于整体的风险评估,要解决“检出限”这个具体的技术问题,还需要参考Q8和ASTM E3263-22。
ASTM E3263-22《制药设备和医疗器械残留物目视检查的合格性标准实施规程》,它主要规定如何通过目视检查来判断制药设备或医疗器械是否清洗干净,也有详细的方法去回应Q7第三个评估方向。
但这里我先介绍Q8,这是更加权威的官方监管文件。EMA在Q8中提到了对目测方法本身的具体要求。比如:光照条件、距离、能否看到所有表面、特定的人员培训、定期视力检查、人员资质考核等。

关于“能否看到所有表面”这个要求需要着重强调一下,我们不妨自问:对于反应罐,通过投料口查看了罐体内部,就算检查完毕了吗?
A:当然不算,很多地方都是“盲区“,比如底阀和管道内部,不借助工具,根本看不到。
B:你说得对,所以我们执行目视检查的逻辑很清晰:在执行前,先划定它的能力边界——目视能把所有表面涵盖的,可以用它;不能被涵盖的,必须要用其他科学手段确定洁净度。
为了回应Q8,简单而言,是做一系列残留量梯度的加标样片,打乱顺序后让多个经过培训的人员检查判断,在特定的光照、距离和角度条件下,判断不同浓度的样片是干净的还是脏的。
传统方法会选择每个人都能识别为脏的、最低残留量的作为目测方法的检出限。但在统计学上,传统方法得出的检出限还不够接近真实值,因为没有考虑实际操作中观察条件的波动和参与测试人员数量的局限性。因此,
ASTM E3263-22
的
方法也给出了数学模型对浓度与检出率进行回归拟合,并给出95%置信度下90%检出率的浓度水平作为目测法的
检出限(VRL),所得到限度会比传统方法高一些,也就是更保守一些。最后把VRL与基于HBEL算出来的单位面积残留限度(MSSR)进行比较,VRL小于MSSR的十分之一,才认为目测方法是可靠的,即需要满足条件:![]() 。
。
VRL(Visible Residue Limit):可见残留限度。
LOD(Limit of Detection):检出限。
在E3263-22中,对VRL作出的定义为——在规定的观察条件下,经培训合格的检查员能够在表面上看到的残留物的最低水平。
在目视检查中,从“检测能力的下限”角度,可以理解为VRL是目测法的LOD,但需要注意,这里的检出限并不是仪器分析意义上的检出限,而是视觉感知的检出限,受人为因素影响大。
总结下来,可以用以下流程图来判断目视方法是否可以单独用在清洁验证,
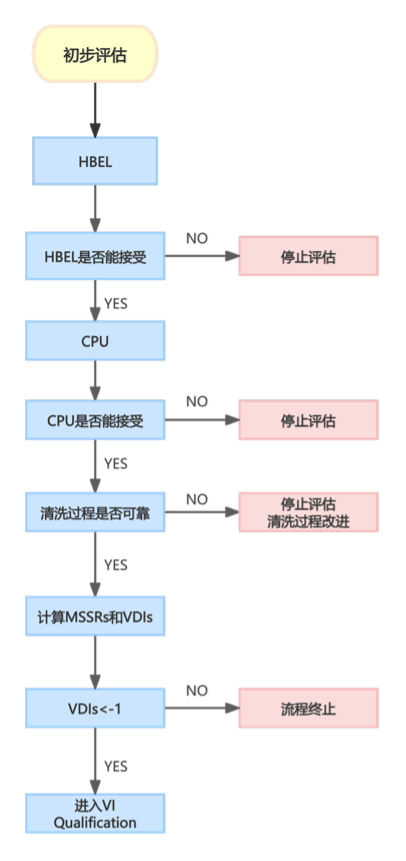
该流程回应了EMA在Q7回答中的三个评估方向:
1、清洁工艺的重现性(Cpu);
2、产品残留的危害度(毒性);
3、目测方法的检出限是否能满足通过HBEL算出来的限度(VDI)。
当然,在评价日常生产时,能否用目测作为日常清洁放行标准,也可以用这个逻辑。
A:做了这么多工作,又评估又试验又统计分析,不如直接都取样检测算了,方便简单又保险,还不用跟检察官解释太多。
B:确实,企业需结合自身业务情况来考虑这一问题。对于产品结构单一或仅从事制剂生产的企业,可能无需在此投入过多精力。
然而,对于一些产品种类丰富的API企业而言,在资源有限的情况下,如果均按最严标准执行,虽最为稳妥,但可能导致资源分散,反而无法在关键风险点上集中投入,造成“捡了芝麻,丢了西瓜”的局面。
此外,从今天讨论到的方法与工具中,我们正可以看到基于风险与科学的质量管理范例。
最后,留给大家一个思考题:以上我们讨论的都是化学残留。如果产品有微生物或者内毒素控制要求,清洁验证还能仅凭目视检查作为唯一标准吗?
参考法规:
FDA《清洁工艺验证检查指南》
NMPA《清洁验证技术指南》
EMA《交叉污染和共用设施清洁限度指南问答》
ASTM E3106-22:关于如何科学地清洗设备
ASTM E3418-23:关于如何计算残留物科学合理限值的标准。
ASTM E3263-22:关于目视检查残留物合格性的标准。
ASTM E3219-20:关于推导基于HBEL的标准
关于我们的工厂验证服务
本文探讨的清洁验证难题,正是gempex德恩咨询日常协助客户解决的真实场景之一。作为源自德国的GMP合规咨询机构,我们在验证与确认领域拥有超过20年的专业积淀,致力于帮助企业将复杂的法规要求转化为可执行的合规方案。我们的工厂验证服务涵盖药品生产全生命周期:
1.验证体系搭建与优化:协助企业建立符合欧盟、FDA、NMPA等国际标准的验证管理体系,涵盖清洁验证、工艺验证、分析方法验证、计算机化系统验证等领域;
2.验证策略咨询与审核:针对具体产品与工艺,提供基于风险与科学的验证策略建议,审核验证文件,确保合规性与可操作性;
3.合规差距分析与整改:对标国内外最新法规指南,全面评估企业验证体系的合规状态,识别改进机会并指导落地实施;
4.验证相关的定制化培训:为企业验证团队提供理论与实践相结合的专项培训,提升内部专业能力。
如果您希望深入探讨验证相关的落地难题,或需要专业支持优化现有的验证体系,欢迎联系gempex德恩咨询的技术专家。我们致力于将严谨的法规语言转化为可执行的企业方案。
如果您想了解更多GMP解决方案,可以访问www.gempexchina.com/gmp-knowledge获取。在那里,您将更深入了解gempex德恩咨询的GMP服务。
如果您有具体的合规难题,请通过以下方式联系我们。
热线:400 166 2002
邮箱:info-cn@gempex.com
关于gempex德恩咨询
gempex德恩咨询深耕GMP合规领域24年,致力于为全球的生命科学企业提供合规、高效及可执行的GMP解决方案,帮助制药、生物技术、原辅包、医疗器械等各方达到NMPA、EU、FDA、PIC/S等GMP标准,减少合规及药品安全风险。
目前,我们拥有60多位经验丰富的GMP专家,全球累计执行项目超过5000个,累计为1000多个客户提供专业服务,业务遍布20多个国家,并与众多知名药企建立了长期的合作关系。

