
#清洁验证 #API #化学工业
近些年来,官方(包括欧盟、美国FDA)对API的生产要求不断增加。在这种情况下,用于生产化学产品中活性药物成分的工厂,在进行清洁时,需要根据经过验证的程序来进行清洁。这也在相应的法规及近些年的检查中,有了进一步的加强,特别是对于生产多产品及共线生产的工厂。
本篇文章来自德恩技术专家Jörg Koppenhöfer编写的《活性成分生产中的清洁验证》,并已发表在ECV(Editio Cantor Verlag)杂志。希望本期分享的(本篇文章,分二期进行)内容对您了解这方面的工作有所帮助!
概括
我们知道,在清洁验证期间,必须提供书面证明,证明定义和描述的清洁程序以可重复的方式实现预期的目的。验证的成功执行,允许在日常操作中,清洁后的取样和分析检查可以减少到必要的最低限度。而关于残留测定验收标准的定义,GMP指南 (第3章和第5章以及附录15) 的修订,以及EMA及PDE指南中提供的新的方法,在此仅简要介绍。
本文主要目的是介绍在化学工业中多产品共线生产活性物质工厂的清洁验证项目的系统实施,并提出关于验收标准确定的方法。
01 指南
说到“清洁验证”这个主题,有很多可供参考的文献[1-4]。很多地方也都提到过清洁验证,但是没有具体说明如何进行清洁验证。其中在这些文献中,PIC/S (PI 006-3) 指南[1]是特别有帮助的。在本篇文章,我们也会多次提及该文件,因为它包含了最详细的指南,特别是可供参考的指南。
在PIC/S指南文件(PI 006-3)中可以看到清洁验证的原则说明,详情见7.1.2、7.1.3和7.1.4[1]。需要特别提及的,在这些内容中强调在清洁验证成功的情况下,日常操作中可以不用取样测试。更多的信息可以查看EU GMP指南附录15[6]。但是本篇内容,在PIC/S指南文件中被提及的更加详细。
02 清洁验证项目的基本程序原则
清洁验证过程项目可分为6个步骤,具体如下图1,且图中每步内容在本文第3-8部分做了详细描述。
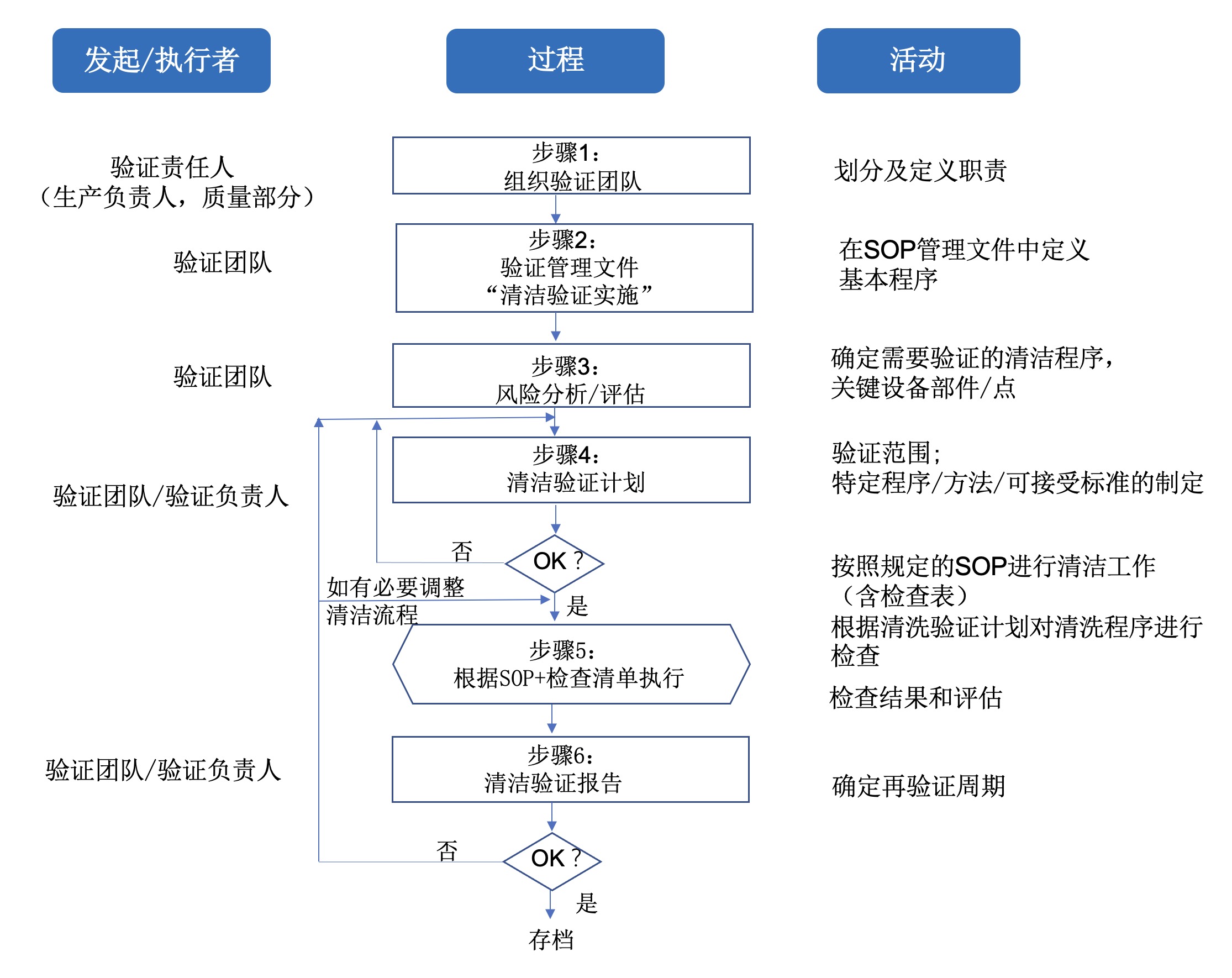
03 步骤1:验证团队
3.1 责任明确
在每个清洁验证项目开始前,组建验证团队并清楚地划分和定义职责。
第一步:明确产品责任问题。根据GMP指南/法规,生产管理部门和质量部门 (Quality Unit,QU) 的管理部门对产品负主要责任。双方负责确保所有GMP要求—包括与生产设施的清洁有关的要求,且在实际运行中得到实施。因此,至少生产管理部门和质量部门共同负责整个验证过程。
在原料药工厂,管理生产的任务通常由相应的工厂经理承担 (见PI 006-3, 2.7.1[1]) 。这些规范通常已经在更高级别的验证主计划中制定。
3.2 验证团队组建
为了管理验证项目的复杂任务,组建验证团队的在实践中是非常有效的。生产管理部门和负责质量的管理部门共同确定负责验证中的特殊任务的人员。对于复杂的项目,项目协调人员在实践中证明是成功的。在验证团队中,需要进一步确定即将进行的验证项目的范围和如何具体实施。验证的所有结果都在团队中讨论和评估。验证团队最终决定验证项目是否成功的。除了验证小组的负责人,以下是小组成员,通常包括质量部门的负责人或其代表。所有的团队成员都被分配了角色和任务(例如,可以参考下文的内容,以及PI 006-3,2.7.4 [1])。
具体来说,活性物质区域的清洁验证项目必须定义并记录以下职位和职责:
负责执行清洁工作的人员(如:工厂领班、管理操作员)
负责清洁过程GMP符合性文件的人员(如:工厂操作人员、操作人员)
负责进行样品和样品分析评估的人员(如:实验室人员)
负责编制结果和准备报告的人员(如:项目协调员或验证协调员)
质量保证部门负责人(如:QU代表)
这些说明通常在上一级验证主计划中制定。
04 步骤2: 概念SOP“执行清洁验证”
在下一步中,验证团队必须确定,在公司是否有一个生效的概念SOP文件指导清洁验证的开展。这个SOP必须描述清洁验证的基本程序,且通常与工厂的所有相关人员协调/培训,以确保程序理解一致避免误解。如果没有这样的SOP,则必须先起草并批准(见PI 006-3, 4.1.1[1])。
概念SOP应该概括地描述基本程序。如图1所示的流程图可能会有所帮助。此外,清洗验证所必需的所有文件/表格/表格模板以及这些文件所要求的内容都应该在SOP中定义或创建。
基本上,标准或资料应提供:
清洗验证过程的一般程序
实施“清洁”风险分析/风险评估的架构、内容和说明
清洁验证计划的架构和内容
在验证必须的文件中包含清洁工作的基本实施
清洁验证报告的架构和内容
05 步骤3: 实施“清洁”风险分析/风险评估
风险分析/风险评估“清洁”是定义特定设备清洁要求的中心文件。验证团队收集并评估所有关键点。应作出下列考虑并记录在案:
工厂(工厂的部分)需要清洁的设备的记录
识别工厂/设备中处理的产品/污染物
确定在工厂/设备中处理的产品/污染物
记录工厂/设备使用的清洁程序/步骤
识别产品和设备组(括号法,最坏情况考虑)
验证范围的最终确定(见PI 006-3,2.5.6和7.3.4[1])
5.1 待清洁工厂/设备的记录
列出整个工厂/设备的清单,并规定工厂每个部分的清洁要求。应注意并记录以下几点:
是否是一个生产多种产品的工厂?哪些工厂/设备用于哪些产品的生产?
工厂部分是否在密闭方式下运行?污染物是否可以从外部进入工厂?清洁的关键点在哪里?
清洁至关重要的点(如死角、难以清洁的区域)。文件可以在图纸的帮助下完成(例如P&I管道图,施工图)
哪些部分需要进行很好的清洁?是否有CIP/SIP要求?
哪种情况下使用哪种清洗剂?
5.2 工厂/设备中处理的产品/污染物的记录
所有可能的输入的物料、产品、清洁剂和其他可能进入各自系统部件的物质也记录在一个清单(物质清单)中。以下几点必须遵守并记录在案:
产生了哪些物质(输入材料、降解/衰变产物、副产物、介质)?
物质的特性(如毒性、功效、溶解度、药效)
关键的质量标准 (微生物、微粒要求)
使用了哪些清洁剂(它们本身可能是污染物吗?它们有什么特性)?
5.3 工厂/设备的清洁程序/步骤的记录
验证小组对整个工厂的所有清洁步骤或清洁程序进行编制和评估。这分为关键和非关键的清理步骤。尤其关键的是多产品车间的产品转换的清洗过程,这些都必须经过清洁验证。
5.4 产品和设备组的形成(最坏情况考虑)/括号法
为了减少验证工作, 让常规的做法更有意义,尤其是防止活性物质,做出以下注意事项/评估(括号法)来提高效率和节约成本的测量和记录这些风险分析/风险评估(见PI 006-3,7.3.5[1]):
设备分组
相应清洗过程的验证结果是否可以转移到其他类似的工厂(类似产品和类似工艺)? 这是可能的,如果清洗过程和相应的系统几乎等同(例如:相同的容器)。
产品分组
在对要清洗的污染物进行性质验证时,是否有可能划分性质相似的产品组?
验证必须只对具有最坏特性(最坏情况)的污染物进行。通常,污染物在所用的清洗剂中的溶解度用于这一目的。然而,在某些情况下,毒性也可能是决定性的。验证计划中产品组形成的文件可以进行,例如,根据表1列出的物质清单。关于活性物质生物技术生产中的清洁验证“ Münsch in Pharm Ind. 07/2018,p.990”这篇文章提供了很好的见解。
5.5 验证范围的最终确定
基于5.1-5.4中的考虑结果,最终在关键系统/设备,和关键污染物的基础上得出需要验证的清洁程序/步骤,结果记录在风险分析/风险评估中。
以上内容为德小恩和大家分享的《活性成分生产中的清洁验证》第一期内容。
参考文献
[1] PIC/S Pharmaceutical Inspection Convention "Recommendations on ... Cleaning Validation. PI 006-3; Sept. 2007.
[2] ICH International Conference on Harmonisation "Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients". ICH Q7. Nov. 2000.
[3] FDA Guide of Inspections of Validation of Cleaning Processes, Jan. 2006.
[4] ZLG Aide Memoire "Inspection of Validation and Qualification in Pharmaceutical Manufacturing and Quality Control**, Chapter 7.
[5] EMA/2012: "Guideline on Setting Health Based Exposure Limits for Use in Risk Identification in the Manufacture of Different Medicinal Products in Shared Facilities (in force since 01.06.2015).
[6] EC GMP Guideline 2015, Annex 15 (Chapter 10.6).
[7] ICH Q3C (R5)/2006 on Impurities: "Guideline for Residual Solvents".
[8] VICH GL18, App.3/1999-Rev.l: "Residual Solvents in ... Active Substances and Excipients.
[9] Allhenn D, Anhalt E. Selection of acceptance criteria for the cleaning validation of multipurpose plants. A position paper of the Pharmaceutical Technical Committee in the BAH. Pharm Ind. 2015; 77(7):1074-80.
[10] Anhalt E. Cleaning validation. Visible clean or analytical detection. Pharm Ind. 2016; 78(1):96-8.
[11] Münsch D. Cleaning validation in biotechnological drug production. Pharm Ind. 2018; 80(7):987-96.

