
#确认4.0 #确认
导读
本文摘自德恩创始人兼中国区总经理Ralf Gengenbach《Qualification 4.0-Unused Opportunities》一文。我们会分为2期进行分享。其中本次分享的是第一部分内容,主要包含确认的发展历程、总结确认效率低下的原因并对每一条提出了建设性的tips总结。
如您想了解和查看英文原文,可点击文末获取原文。
作者简介

Ralf Gengenbach
gempex创始人、CEO
GMP资深顾问
ECA协会主席
VIP3000主席
1987年毕业于德国卡尔斯鲁厄大学 (TU Karlsruhe)化学工程专业,硕士学位。
1987年–1997年,就职位于路德维希港市的BASF总部,负责生物技术生产原料药的工艺开发工作。
1997年–2002年,任职于DIS AG,建立质量咨询事业部并担任事业部负责人。
2002年,在德国曼海姆创立gempex。
作为多个协会工作委员会的成员,Ralf参与了多个GMP法规的制定和指南的协调工作,与此同时他仍然作为一名顾问参与全球具有重大挑战性的GMP项目,他也是一位第三方审计官。自2013年以来,Ralf Gengenbach一直担任VIP 3000的主席。他常年活跃于行业内各大协会的专家委员会进行讲座,同时也是诸多出版物的作者和演讲人员。他撰写的《GMP—原料药的确认与验证》一书被誉为是业界标杆之作。
↓↓↓
1987年,美国食品药品监督管理局(FDA)通过了“工艺验证原则”[1]奠定了验证的基础,其中也包含设备确认。但后者仅限于“安装确认”(IQ)。
多年以来,药品检查公约(PIC)(现在扩展为国际药品检查合作组织(PIC/S))在确认的概念中增加了“运行确认”(OQ)、“性能确认”(PQ)和后来的“设计确认”(DQ)等要素。
然后又是30年过去了,这个30年,制药行业一直被该话题以及相关大量的文件所困扰。这也推动了美国材料与试验协会 (ASTM)[2]形成了一份标准文件,该文件希望利用良好工程管理规范(GEP)来简化该程序。在工业4.0的时代,制药行业陷入了停滞不前。确认因其不受控的时间和成本已发展成为项目的“拦路虎”。本文旨在介绍当下确认的基本问题,阐明问题的由来,并且提供如何高效地实施确认的建议。
01 确认的重要性及其发展
20世纪80年代引入的确认概念、其基本理念是:质量不是检验出来的,而是必须通过药品生产涉及到要素的有序和有效性来保证其质量。除去训练有素的人员、良好和可追溯的文件、符合质量标准的原材料、详细的规程等,还要求要有经过正确安装和运行的技术设备。应当确保通过制定好的检查清单对正确安装和运行的系统进行确认。将质量部门整合到确认中,来对相应的测试计划在执行前进行正式批准并最终评价偏差和报告,强调了确认的重要性。
确认中安装和运行确认的要素进一步得到补充,先是性能确认,然后是设计确认。其中的设计确认是因为大家认识到质量是在早期设计阶段就决定了上限。在选用了错误设计的情况下,安装确认最多只能证明技术设备按照错误设计进行的建造和安装符合错误的设计标准,但是错误本身无法被纠正。
随着确认进一步的发展,大家希望摆脱纯粹正式的核对清单的填写,而更多注意关键的方面。于是就引入了风险评估的主题。此外,还引入了质量关键属性(CQA)和质量关键工艺参数(CPP),在确认过程中应特别关注CQA和CPP的识别和确定。
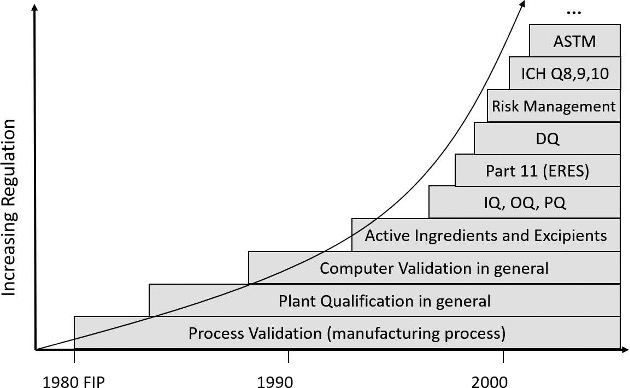
图1:确认的法规发展(数据源:作者/gempex GmbH)
02 确认受到质疑以及被重新定义
当确认在许多企业造成堆积如山的文件、无休止的形式主义、项目延误和巨大的额外成本,却在实际上没有展现出来任何明显可观的好处后,批评的声音于是越来越大。
2007年,ASTM针对制药和生物制药工厂推出了相应的指南。文件ASTM E 2500[3]规范包括控制和自动化系统在内的设备的规格说明、设计和确证(确认)。除了基于风险和科学方法,CQA和CPP的阐述以及面向“质量源于设计”(QbD)的方法等已知主题之外,现在也将“主题专家”(SME)和供应商更多的参与活动考虑在内,并通过所需的持续流程改进来完成。
简单来说,ASTM标准的建议也被翻译为“回归本源”——合理、可靠的工程工作,以及只有必要时(最后阶段)质量部门才参与。
03 确认效率低下的原因
1. GEP或工程技术的再创新
在与工程技术相互作用的确认中也观察到类似的现象。起初,它是简单的安装确认(IQ)和运行确认(OQ)检查清单。但今天已大大扩展到包括用户需求标准(URS)和功能/详细设计说明(FDS,DDS)、工厂验收测试(FAT)和现场验收测试(SAT)文件,测试计划以及更多的内容。这些文件日益模糊了工程技术和确认之间的界限。虽然URS、FDS、DDS、FAT和SAT等术语已经存在很长时间了,但今天它们被用于与GMP和确认相关的内容中,就好像它们就是为此而发明的一样。
原则上,如果没有与质量部门的相互影响,没有GMP/确认的需要导致相关文件需要以某种形式展现、需要用到特定签名以及需要执行变更控制,这都不会是一个悲剧。如果FAT或其他技术文件由质量部门进行检查,就会产生这样的问题,质量部门是否有能力评估这样的文件。另一方面,它大大增加了形式上的要求从而增加了相关的工作量。整体而言,越多属于工程技术的活动和文件被纳入确认中时,确认过程就会变得越复杂和耗时。这并不意味着在描述这类文件的范围和内容时,把质量部门排除在外。但是最终的协调、调整和批准都应是“主题专家”的责任。
Tip小贴士
应确保明确区分GEP和GMP文件和活动。只有真正相关的和确认特定的文件,才应该包含在正式的确认概念中,其他文件尽可能留在工程部分。
2. 验证团队——人人都有意见,人人都能做决定
GMP的一个基本特征,特别是确认的一个基本特征,就是工作需要跨部门来完成。跨部门活动以及对信息数据需求的协调都很不容易,而更困难的是文件的协调和最终发布。因此,在这里,我们建议:对项目组织进行早期和明确的定位,以及对必要的工具和仪器的定义,这都是确认工作成功的基础要素。详细的建议内容部分,可以大家点击文末,阅读原文。这里,此处仅做概括展示。
Tip小贴士
主要的关注点应该放在项目的早期组织。确认项目是高度复杂和跨部门的。一般应该按照“每个人都可以发表自己的意见,但只有一个人决定”的原则进行。
3. 用户需求标准(URS)的问题
URS在技术变更管理方面也很重要。即使在正在进行的建设项目中,也应该讨论、评估和记录重大的变更,特别是GMP要求相关的变更。如变更情况存在时,除另有规定外,应基于URS中的标准来做出决定。
一份过于详细的技术性的URS会将技术性要求、但不是对于确认必要的相关内容掺入到确认活动中,因此使URS的处理成为一个真正的挑战。
图2:计划过程中的需求标准和功能说明
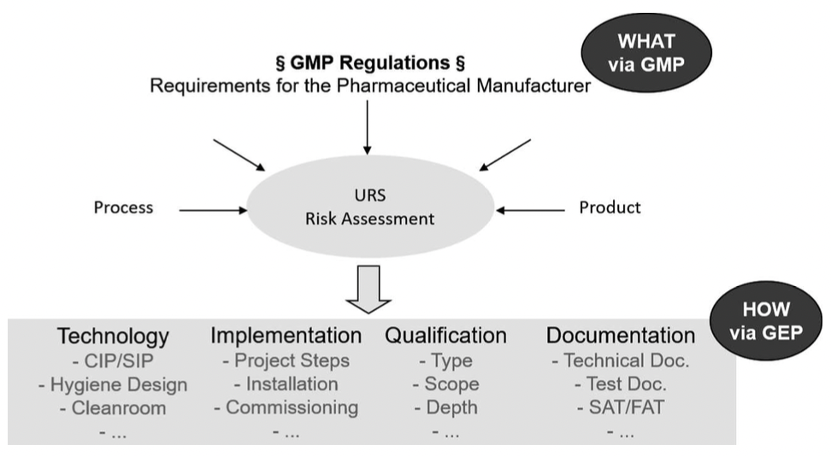
图3:根据URS得出技术规范
URS应专门关注用户——系统未来的使用人员。他想生产哪些产品?面向哪些市场?考虑哪些法规?有什么特殊的挑战?——良好的可清洁性、多产品共用厂房、关键产品。哪些控制和监控参数需要发挥作用?哪些质量属性关键的?然后工程师的任务就是从这些用户要求中得出技术规范。
Tip小贴士
在确认的概念中,应该清楚明确地定义什么是用户说明,什么是技术规范,以及由谁在哪个层级上创建这些文件。用户需求标准(URS)应该只包括用户角度出发的需求,而技术细节应该由工程师编制在技术规范中。
以上内容是我们摘自Ralf Gengenbach撰写的《Qualification 4.0-Unused Opportunities》第一部分。后续会分享第二部分--有关确认的规划、实施等内容,敬请关注!
参考文献
[1] FDA, Guideline on General Principles of Process Validation, May 1987.
[2] Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment, ASTM 2500-07, revised 2013.
[3] Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment, ASTM E 2500, Aug 2007, revised version 2013.

