
400-166-2002
#MAH #药品上市
MAH(药品上市许可持有人)是近期行业内密切关注的热点,MAH制度使上市许可与生产许可分离管理,并对药品的全生命周期及整个供应链全面负责。
那么,上市许可持有人可以自行研发持有、可以各项工作委托持有、可以获得授权持有等等,那么应该怎么部署相关工作呢?通常我们应该首先定义好商业模型,根据各自的职责分配工作,搭建质量体系,并进行良好的管理和实施。
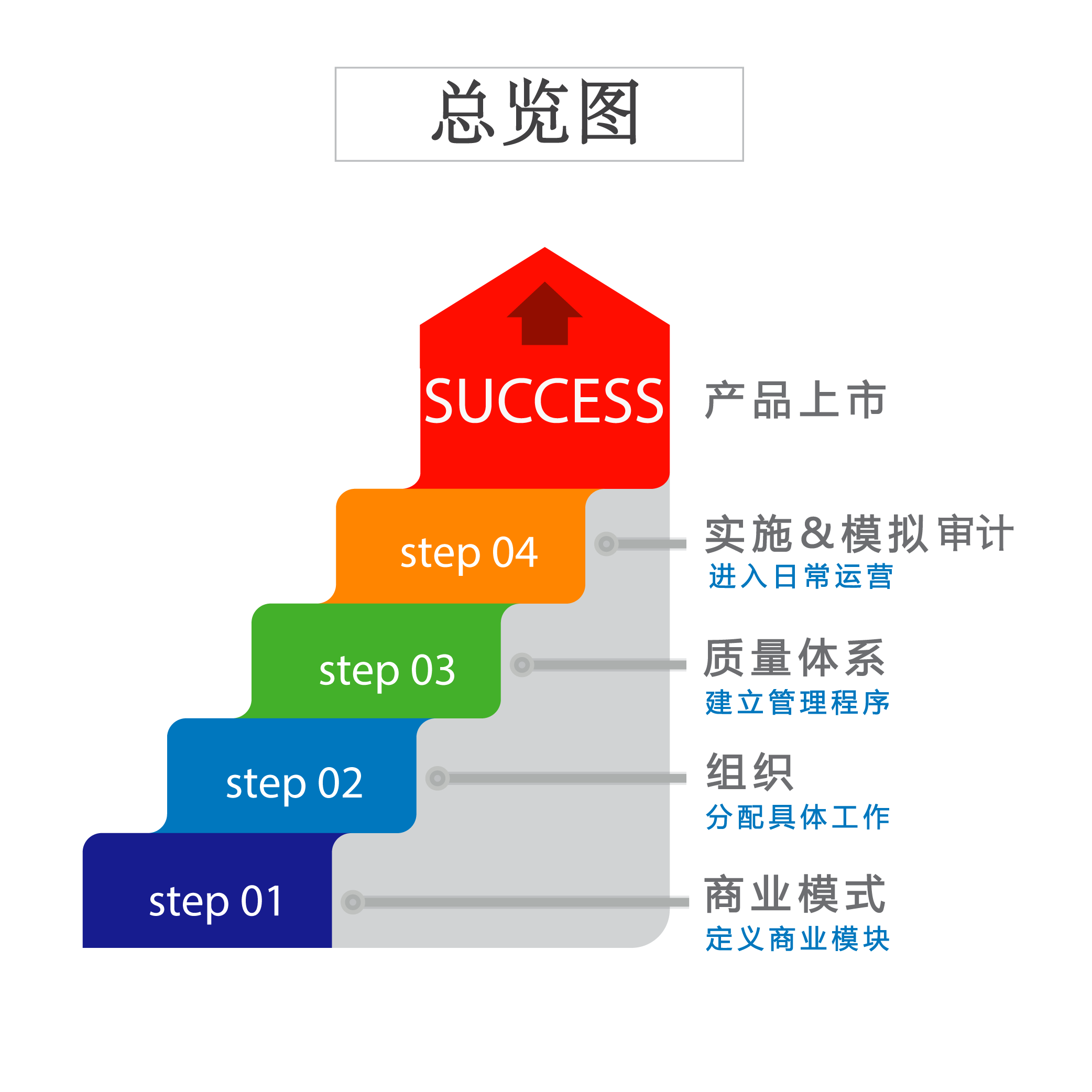
01 定义商业模式
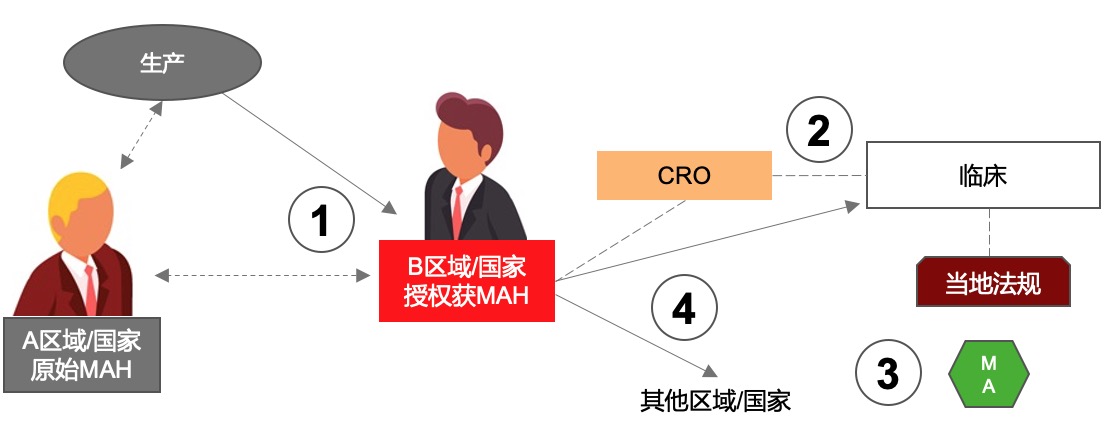
在通过授权获得的MAH的商业模型,如何对模型简洁清晰的勾勒关系呢?见上图所举例的一种商业模型。
定义好商业模型后,我们需考虑的要点:
1. 生产 & 供应 (主要由CMO提供)
如何运输 (A港口 → B区域 / 其他国家)
如何储存 (在B区域 / 其他国家)
如何实施来货检验及放行
2. 临床试验(由CRO提供)
如何管理 (TMF – 试验主文件)
如何盲法试验管理(由承包公司或者外包提供)
3. 上市许可申请 (可选择外包)
法规/信息更新处理
与官方监管机构/外包方的沟通
4. 发运 (可选择外包)
如何发运至客户
1. 运营 (包括 QA)
供应商资质确认和审计
维护质量协议
监督生产&供应及发运的活动
产品的放行
2. 商业运营
专注市场、客户和机构的关系
上市许可申请
投诉、召回等
3. 临床运营
专注于临床试验相关主题活动
03 质量体系的搭建
第一步:MAH必须具有合适的管理体系,以确保符合GMP的生产、GDP的运输和发运、放行、投诉和召回、药物警戒GVP等。注意检查以下主题文件:
1. 公司内部常规程序
组织架构/ 职位说明 / 培训
常规文件管理/存档/归档/数据完整性
采购系统及相关文件
质量评审和自检程序
......
2. 生产&分销
供应商评估和确认
供应商和服务提供商的审计,包括定期审计(例如原料药、制剂生产商、承包商、运输、存储、实验室和任何其他外部服务,如质量保证)
合同管理,包括质量协议
重新包装及重新贴签,包括标签放行
进口后用于上市的最终产品放行
产品接收方的数据管理
如果由MAH负责运输,则需做相关运输验证
…...
3. QA & 监督
偏差/变更/投诉管理的最低要求(对产品生产商的投诉)
年度报告管理
药物警戒系统
审计和检查的准备和管理(这里指MAH被审核或检查)
第二步:因为临床试验的特殊性,它将被视为一个完整的独立系统,同时也是考虑到所需的SOP。虽然由于活动将外包给CRO并大多数SOP可由他们提供,但MAH将承担一些职责,例如至少MAH将作为申办方。所以,需要做以下临床试验的程序职责:
组织、角色、职责
(谁是申办方,谁应该负责什么)
评估和CRO的资质确认
CRO的审计
与CRO达成的质量协议
试验主文件和其他文件的管理
盲法试验程序和职责
临床试验药品的进口和放行
定期的结果报告系统
…
第三步:对于上市许可申请,MAH实际上并不需要那么多的SOP,但他们须了解并遵守上市国家的法规要求及程序,可能也会使用专业的注册代理。所以,在SOP中,主要涵盖以下内容:
许可申请相关的职位和职责
许可申请数据的管理和归档
变更和版本控制 / 许可申请更新程序
与官方监管机构的交流
…
最后,我们还得做完上市许可申请的活动,例如:
药品生产许可证
通过eCTD形式将文件传递给官方监管机构

