
400-166-2002
2026-05-28
上期我们分享了《中国药企出海GMP合规生存指南》,梳理了FDA、EMA、PIC/S等海外监管趋势。
本期我们聚焦Biotech企业——尤其是临床前、I/II期的早期公司——拆解GMP合规在融资、BD交易与出海中的阶段性要求与实践逻辑。

中国Biotech正在以前所未有的速度融入全球创新药产业链。2026年以来,国产创新药BD项目数和披露金额的全球占比分别达20%和75%。
但亮眼的数字背后,另一组数据值得警惕:动脉网统计显示,2020年完成的62起License-out交易中,40%已明确终止合作。这意味着,签单只是起点,真正的考验在协议签署之后。在诸多导致交易终止的原因中,CMC与GMP合规问题正在成为一个越来越关键、也往往被早期团队低估的因素。
GMP的法律强制性:
没有“豁免期”
很多Biotech认为GMP是“商业化阶段才需要操心的事”,但监管的趋势正在将合规压力推向更早的环节。
2026年生效的新版《药品管理法实施条例》进一步强化了全生命周期监管,其第八十二条明确要求:从制备I期临床样品开始,制备过程即应符合GMP要求。
但“强制性”不等于“同等严格程度”。监管体系认可阶段适配性原则,即GMP的要求随产品开发阶段递进。ICH Q10指出,质量体系的设计应反映产品生命周期的不同阶段;PDA第56号技术报告(2026年修订版)也强调,早期阶段的GMP重点在于数据完整性和工艺的可追溯性,而非商业化所需的全面工艺验证。
因此,Biotech面临的问题不是“要不要做GMP”,而是“在每个阶段做到什么程度”。
各阶段GMP的实操基准
基于NMPA、FDA及ICH指南,结合行业通行实践,各阶段GMP合规的核心要求可概括如下:
临床前/早期开发阶段
Pre-IND/Phase 0
I期临床阶段
II、III期临床阶段
商业化阶段
BD交易中
GMP合规的具体影响
在BD交易(尤其是License out)的技术尽调中,买方通常会从以下维度评估卖方的GMP状态:
数据完整性
审计人员会追溯原始数据(电子记录与纸质记录),检查是否存在后补记录、审计追踪未开启等常见问题。数据完整性缺陷在FDA警告信中常年位居前三,同样也是MNC买方最敏感的否决项。
与CDMO的权责边界
若采用外包生产模式,买方会重点审查Biotech与CDMO之间的质量协议是否清晰定义了变更通知义务、审计权利、偏差处理流程等。实务中,大量Biotech使用CDMO提供的模板协议直接签署,导致在关键变更发生时未被及时告知。
阶段适配性是否合理
买方不会要求I期项目具备商业化级别的验证包,但会评估:现有GMP体系是否为其所处的开发阶段提供了足够的控制?如果存在“过度宽松”或“过度僵化”的情况,都可能影响买方对团队成熟度的判断。
目标市场的GMP标准差距
对于拟进入欧美市场的产品,买方会评估当前质量体系与FDA/EMA标准的差距。例如,是否按ICH Q7管理原料药?无菌保障是否符合EU GMP Annex 1?这些差异直接转化为交易后的补救成本,进而影响估值。
出海:
GMP合规作为准入门槛
不同目标市场对GMP合规的接受规则存在显著差异,但共同趋势是趋严。
中国监管层也在主动推动GMP国际接轨。2025年发布的《出口药品生产监督管理规定》征求意见稿明确提出“促进药品出口贸易”,鼓励企业按照国际标准建立质量体系。这意味着,未来国内GMP符合性检查的标准将逐步向PIC/S靠拢,企业如果提前布局,将获得双重便利。
对Biotech的实操建议
基于上述分析,有几点判断供从业者参考:
第一, 早期融入GMP思维是“省大钱”的策略。在一款创新药处于临床I期甚至更早的阶段时,提前建立符合国际标准的GMP意识和基础质量体系,远比在项目进入临床后期甚至准备出海申报时才发现早期记录数据完整性不足、工艺稳定性记录缺失等问题要经济得多。
第二, 分阶段建立GMP体系,适配而非过度。借鉴“阶段适配性”的理念,不必在早期就用商业化标准来束缚团队。临床前阶段更多是理念培训和数据记录规范,I期临床正式引入GMP但保持一定灵活性,随着临床阶段的推进逐步加严各项要求。这样既不会扼杀创新,也能确保产品在进入关键临床阶段时质量体系已基本就绪。
第三, 出海前务必完成GMP差距分析。不同目标市场对GMP合规的要求差异很大。新加坡PIC/S、欧盟GMP、美国cGMP各有侧重,差距分析可以帮助企业明确“当前在哪、目标在哪、如何到达”,避免带着合规盲点上路。
第四, BD交易中GMP尽调不可缺席。无论是作为交易方寻求License out,还是作为买方评估管线,GMP合规都应该是技术尽调的核心内容之一。提前做好合规准备,不仅有助于提升交易估值,也能加速交易进程。
gempex德恩咨询能为您做什么?
Biotech团队通常精简,资源有限,管线推进压力大,多数依赖CDMO生产,内部可能没有专职的QA或CMC合规团队。在这样的现实约束下,建立一套“够用但不浪费”的GMP体系,同时满足BD交易和未来出海的合规预期,是一项现实挑战。
针对Biotech的特殊需求,gempex德恩咨询提供以下可弹性配置的服务:
为什么选择gempex德恩咨询?
我们深耕GMP合规领域24年,60余位专家覆盖NMPA、FDA、EMA、PIC/S、WHO等主流监管体系,全球累计执行项目超过5000个。
我们不提供“模板化”方案,而是基于您的管线阶段、团队规模和融资节奏,输出可落地、分阶段、不浪费一分钱的GMP合规服务。
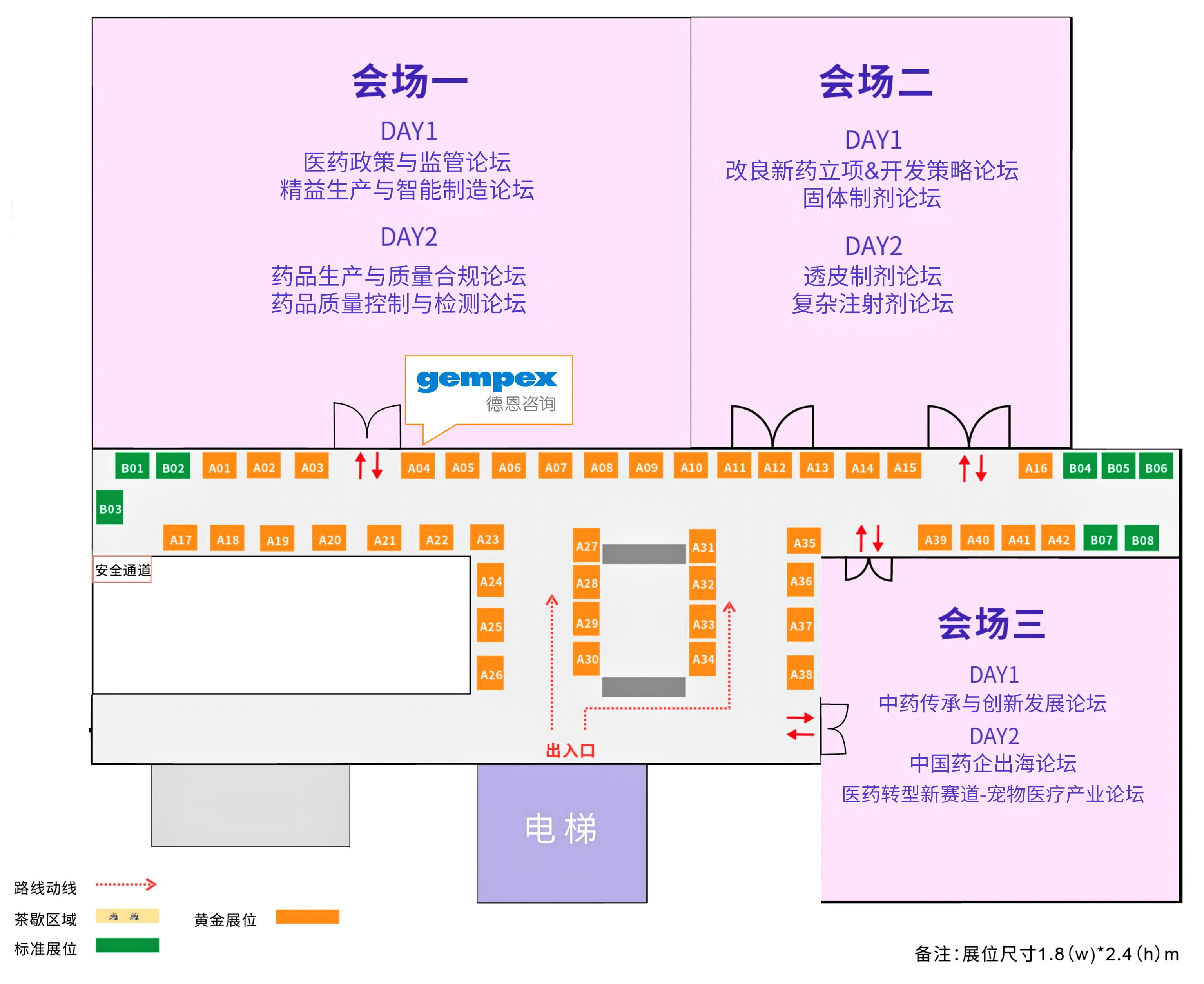
GMP合规的落地细节,一次面对面的沟通往往胜过千言万语。6月2-3日,成都CPI·西南制药工业大会,欢迎来我们的展位【A04】聊聊。
如果您想了解更多GMP解决方案,可以访问www.gempexchina.com/gmp-knowledge获取。在那里,您将更深入了解gempex德恩咨询的GMP服务。
如果您有具体的合规难题,请通过以下方式联系我们。
热线:400 166 2002
邮箱:info-cn@gempex.com
关于gempex德恩咨询
gempex德恩咨询深耕GMP合规领域24年,致力于为全球的生命科学企业提供合规、高效及可执行的GMP解决方案,帮助制药、生物技术、原辅包、医疗器械等各方达到NMPA、EU、FDA、PIC/S等GMP标准,减少合规及药品安全风险。
目前,我们拥有60多位经验丰富的GMP专家,全球累计执行项目超过5000个,累计为1000多个客户提供专业服务,业务遍布20多个国家,并与众多知名药企建立了长期的合作关系。

